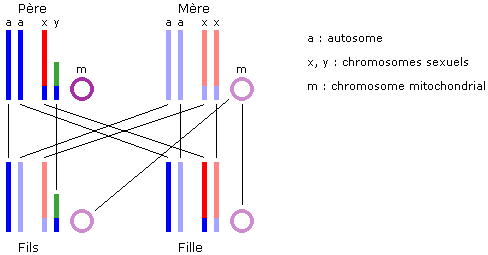
Conseils spécifiques ŕ ce devoir :
L'introduction du sujet présente la maladie comme étant due ŕ la mutation du gčne de la phénylalanine hydroxylase, elle fournit un renseignement important selon lequel une personne sur 63 est porteuse du gčne. Cette seule information permet déjŕ de prédire la localisation autosomale du gčne et le caractčre récessif de l'allčle muté car :
(une personne sur 63 est hétérozygote, dans la descendance d'un couple hétérozygote pour un gčne autosomal, 1/4 de la descendance est homozygote pour l'allčle muté du gčne).
La nécessité de connaître la réponse ŕ la premičre question pour faire la seconde partie du devoir impose le plan ŕ adopter. J'ai choisi de traiter la seconde question du sujet dans deux parties séparées. La difficulté de la troisičme partie réside dans le fait que les génotypes des parents ne sont pas certains.
En conclusion, on attend bien sűr une réflexion sur l'utilisation des connaissances sur l'hérédité pour prédire le risque de maladie génétique dans la descendance d'un couple. Cette réflexion doit aboutir aux moyens modernes de prévenir les maladies génétiques (diagnostic prénatal voire préimplantatoire et interruption volontaire de grossesse).